
EU IVDR
NEXT EPISODE
In case you’ve missed the previous episodes, find them all here!
Episode 5
Episode 4
Episode 3
Episode 2
Episode 1
Trailer
We are committed to keep you informed about the changes in our portfolio planned for IVDR compliance.
Fill out the form below to get regular emails on our journey!
Sailing smoothly through the challenging waters
of the EU IVD Regulation (2017/746)
The Regulation (EU) 2017/746 on in vitro diagnostic medical devices (IVDR) represent a challenge for all of us. But where there is a challenge, there is always an opportunity!
This is our chance to meet the challenge together and work in partnership to ensure a smooth transition to IVDR compliance.
With the positive changes that the MDR and IVDR will bring to the European regulatory framework, we look forward to having our products and unique technologies continue to serve you and your patients.
The EU IVDR – What does it hold for Health Institutions ?
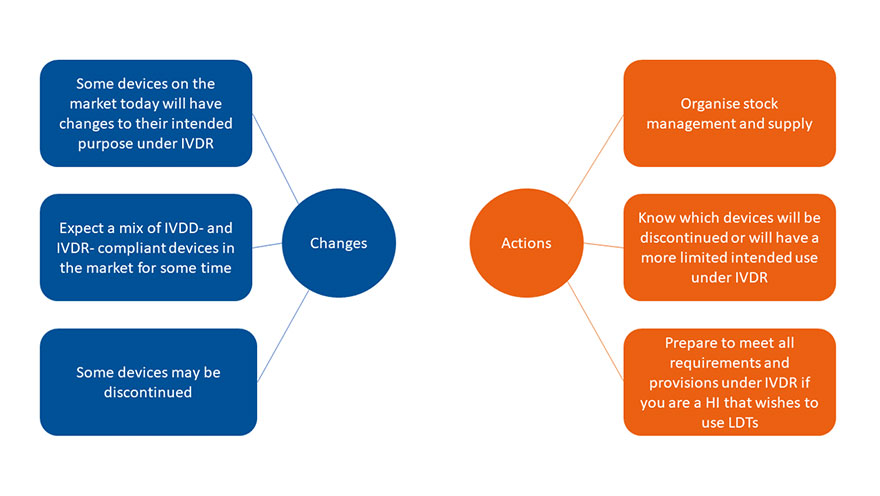
The EU IVDR affects all IVD medical devices and will bring about several changes. Manufacturers are reviewing their product portfolio, and certain devices may be discontinued.
Health Institutions (HIs) should be aware which of their products will be affected and prepare ahead to ensure continuity after the IVDR date of application.
LDTs (Lab Developed Tests) are in scope of the IVDR as well, which means that the LDT needs to meet certain requirements and the health institution can only use the LDT if certain conditions are met.
Planning and preparedness are key!
What is the IVD Regulation about?

Introduction
The EU In Vitro Diagnostic Medical Device Regulation 2017/746 (IVDR) took effect on 26 May 2017, and a transitional period of 5 years is in force till 26 May 2022.
The IVDR replaces the EU's current Directive on In Vitro Diagnostic medical Device (IVDD) (98/79/EC) to ostensibly ensure a higher level of health and safety for making available and putting into service devices in the EU market and setting new rules for applying a CE mark to IVDs. Being a regulation, it does not need to be transposed into the National Law and hence ensures uniform interpretation throughout the Union.
Objectives of the IVDR
Patient safety
To ensure a high level of human health and patient safety
Free & fair trade
To ensure smooth functioning of the internal market
Innovation
To provide a regulatory framework that is supportive of innovation and competitiveness of the European medical devices industry
Harmonisation
The regulation must be implemented in its entirety by all members states of the EU.
Lab developed tests (LDTs)
How will the IVDR impact LDTs?

Why are LDTs under the scope of the new IVDR?
LDTs are now governed by the IVDR.
Rules for the use and manufacturing of LDTs have now been established under the IVDR to ensure the highest level of protection, which is one of the main objectives of the IVDR.
No requirements are described for LDTs in the existing IVD Directive (98/79/EC).

HIs wanting to use their LDTs must consider these rules
- Development and use of an LDT is possible only in the absence of a CE marked alternative or if the performance of the equivalent CE marked device is not sufficient for the target patient group
- HIs/Laboratories must adhere to Article 5(5) and the LDT must meet the general safety and performance requirements (Annex I) of the IVDR
- The EU Member States will oversee LDT devices manufactured and used within the healthcare institutions in their territory
Rules for Health Institutions
Key requirements for Health Institutions (HIs)

Manufacturing and using LDTs- Key requirements for Health Institutions
The LDT shall be manufactured and used within framework of a Quality Management System
The HI shall be accredited according to EN ISO 15189 (or alternate national provisions)
The LDT must comply with General Safety and Performance Requirements (GSPR – Annex I of the IVDR)
The HI will review experience gained from clinical use and takes the corrective actions, where necessary
The HI will compile documentation on device manufacturing, design and performance
The HI will make a declaration of conformity publicly available

Documentation requirements per Article 5(5)
Health Institutions should prepare documentation that describes:
- The intended purpose of the device
- The manufacturing facility
- The manufacturing process
- Device design and performance data in relation to its intended purpose
The documentation should be of sufficient detail to demonstrate that all applicable GSPR as outlined in Annex 1 are met.
The IVDR prescribes the above requirement for class D devices. However, the Regulations allows Member States to apply this provision also to class A, B or C devices.
Health Institutions and IVDR readiness
Know that preparation is key!
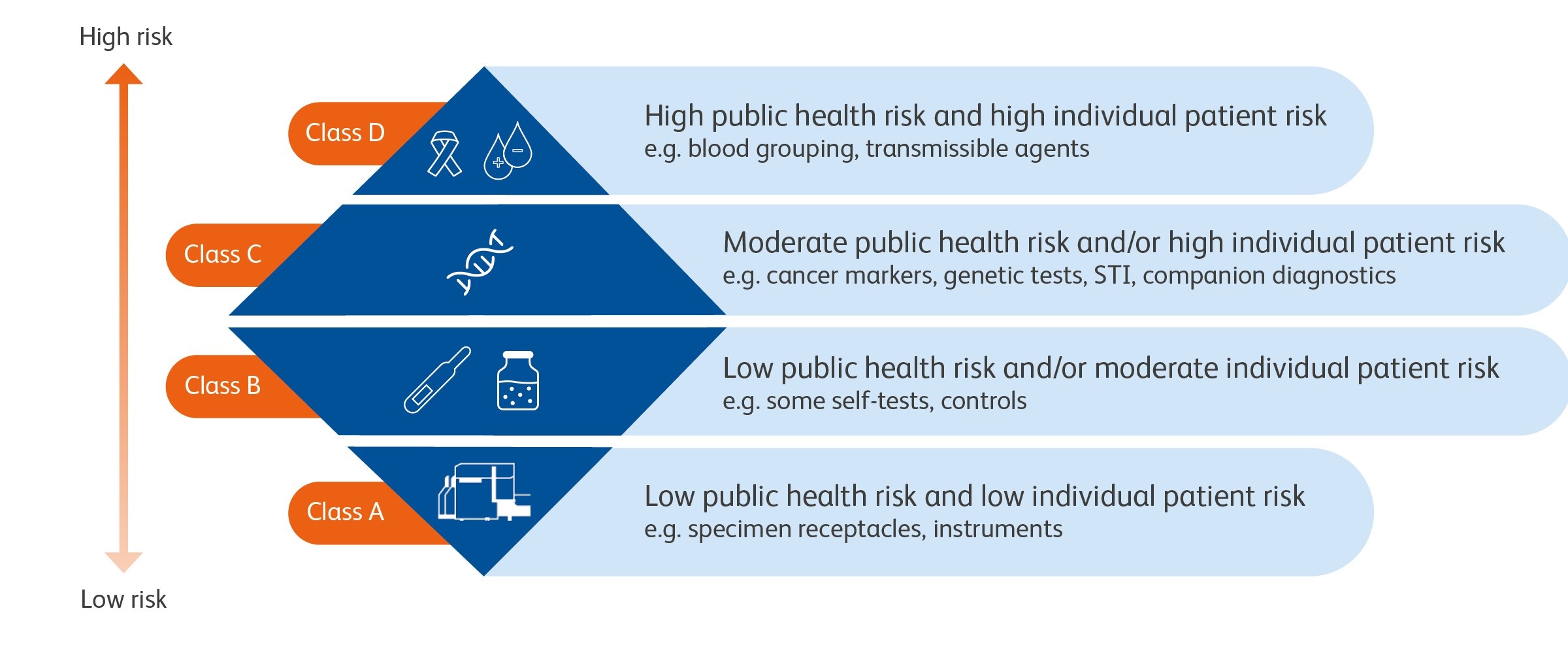
New classification and requirements under IVDR
New risk-based classification system
New conformity assessment
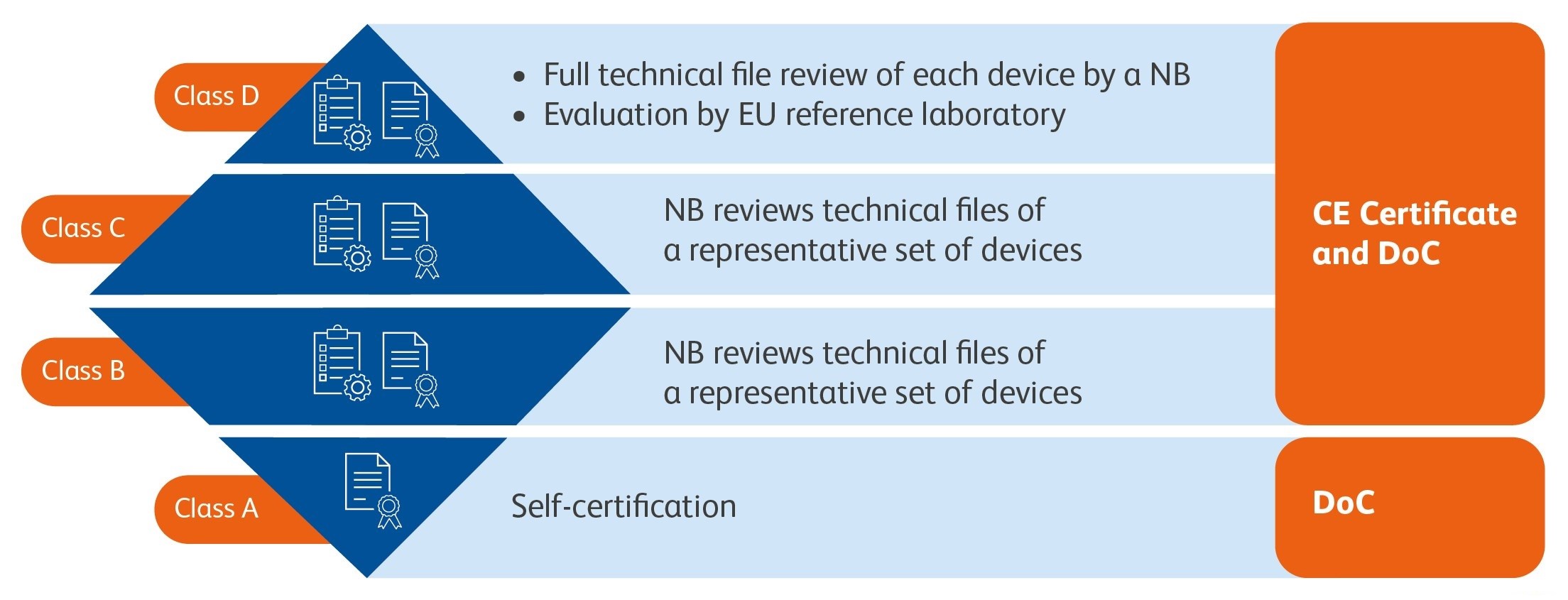
NB ( Notified Body) = Notified body is a third-party certification body that conducts conformity assessments for those devices that are not self-certified. If compliance is demonstrated, the CE-certificate will be issued by the notified body and the manufacturer can draw up the DoC
DoC (Declaration of Conformity) = The DoC serves as a confirmation that all applicable CE-marking legislations are met for the listed devices.
For manufacturers, the IVDR foresees a conformity assessment route for each risk class as a prerequisite for the Declaration of Conformity, and where applicable the CE-certificate.
Class C and D additional requirements
There are additional requirements for Classes C and D
Reports for each Class of Device
- Summary of Safety and Performance (SSP)
- Periodic Summary Update Report
Surveillance Assessment
- Annual, by Notified Body to ensure manufacturers apply the approved Quality Management System (QMS) and Post Market Surveillance (PMS) plan
- Notified Body can visit critical suppliers or subcontractors
Unannounced audits by a NB
- At least every 5 years
- NB can test devices at site and/or from market
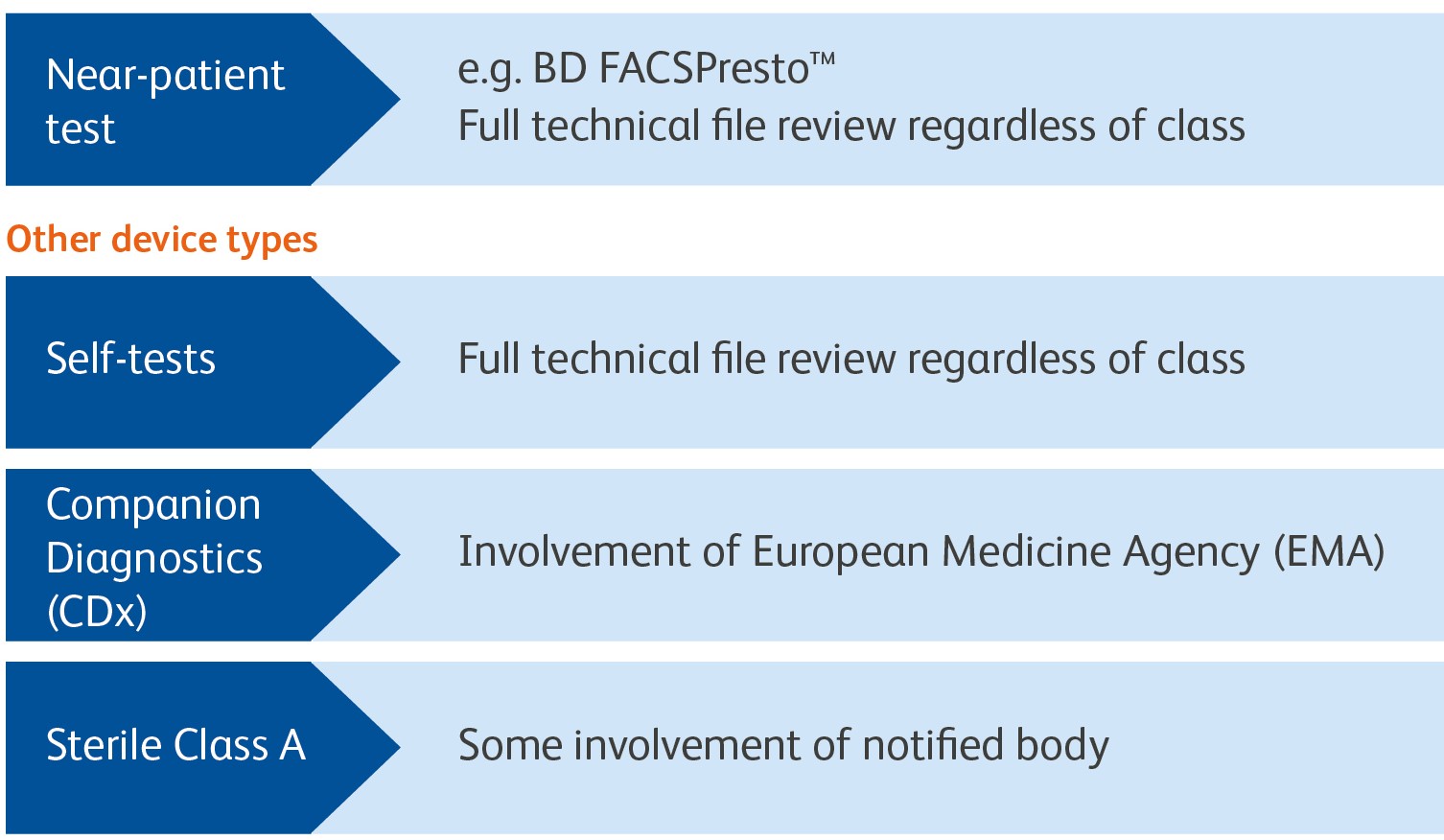
Additional considerations for specific device types
New conformity assessment for specific device types

Traceability and Transparency
One of the key aspects to achieve the IVDR objectives
Enhanced levels of transparency and traceability with IVDR

The CE marking

Declaration of conformity
- Is the basis of CE-marking
- Is the document in which the manufacture declares conformity of the devices to all applicable CE-marking legislation
New requirements for the DoC document compared to IVDD:
- Includes SRN - Single registration number*
- Includes the BUDI-DI
- Translation into an official EU language required by the Member States
* SRN is a unique number that will be assigned to the Economic Operator [Manufacturer, Authorized Representative, Importer] after registering in EUDAMED.


