
Nouvelle réglementation IVDR
Vous avez raté les épisodes précédents? Retrouvez-les ici :
Episode 5
Episode 4
Episode 3
Episode 2
Episode 1
Trailer
Naviguer en douceur dans les eaux troubles de la réglementation européenne pour les DIV (2017/746)
Le règlement 2017/746 sur les dispositifs médicaux de diagnostic in vitro (IVDR) représente un défi pour nous tous. Mais là où il y a un défi, il y a toujours une opportunité !
Nous avons la chance de relever le défi ensemble et de travailler en partenariat pour assurer une transition en douceur vers le respect de la directive sur les dispositifs médicaux de diagnostic in vitro.
Avec les changements positifs que le Règlement relatif aux Dispositifs Médicaux (MDR) et l'IVDR apporteront au cadre réglementaire européen, nous espérons que nos produits et nos technologies uniques continueront à vous servir, vous et vos patients.
Nouvelle Réglementation IVDR - Que prévoit la réglementation pour les établissements de Santé?
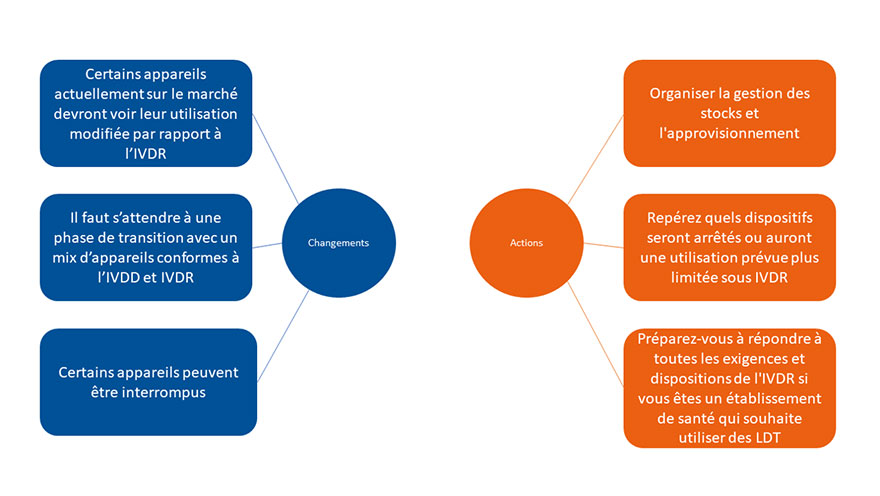
Le Règlement européen sur les dispositifs médicaux de Diagnostic in vitro concerne tous les dispositifs médicaux de Diagnostic in vitro et entraînera plusieurs changements. Les fabricants sont actuellement en train de repenser leur portefeuille de produits, et certains produits pourraient être abandonnés.
Les établissements de santé doivent être au fait des produits qui seront ciblés et se préparer à l'avance pour assurer la continuité après la date d'application de l'IVDR.
Les LDT (Tests Développés en Laboratoire) font également partie du champ d'application de l'IVDR, ce qui signifie que les LDT doivent répondre à certaines exigences et que l'établissement de santé ne peut utiliser les LDT que si certaines conditions sont remplies.
La planification et la préparation sont essentielles !
Quel est l'objet de la réglementation IVDR ?

Introduction
Le règlement 2017/746 sur les dispositifs médicaux de Diagnostic In Vitro (IVDR) est entré en vigueur le 26 mai 2017, et une période transitoire de 5 ans est en vigueur jusqu'au 26 mai 2022.
Le règlement sur les Dispositifs Médicaux de Diagnostic In Vitro (DMDIV) remplace la directive actuelle de l'UE sur les dispositifs médicaux de diagnostic in vitro (98/79/CE) afin de garantir un niveau plus élevé de santé et de sécurité pour la mise à disposition et la mise en service de dispositifs sur le marché de l'UE et établit de nouvelles règles pour l'application d'un marquage CE aux DMDIV. Étant un règlement, il n'a pas besoin d'être transposé dans le droit national et garantit donc une interprétation uniforme dans toute l'Union.
Les objectifs de la réglementation IVDR
Sécurité
patients
Assurer un niveau élevé de sécurité des patients en matière de santé humaine
Commerce libre et équitable
Assurer le bon fonctionnement du marché intérieur
Innovation
Fournir un cadre réglementaire qui favorise l'innovation et la compétitivité de l'industrie européenne des dispositifs médicaux
Harmonisation
Le règlement doit être mis en œuvre dans son intégralité par tous les États membres de l'UE.
Tests Développés en Laboratoire (LDT)
Quel sera l'impact de l'IVDR sur les LDT ?

Pourquoi les LDT sont-ils concernés par le nouveau règlement ?
Des règles pour l'utilisation et la fabrication des LDT ont été établies dans le cadre de l'IVDR afin d'assurer le plus haut niveau de protection, ce qui est l'un des principaux objectifs de la réglementation.
Aucune exigence n'est décrite pour les LDT dans la directive IVD existante (98/79/CE).

Les institutions souhaitant utiliser leurs LDT doivent tenir compte des règles suivantes :
- Le développement et l'utilisation d'un LDT n'est possible qu'en l'absence d'une alternative marquée CE ou si les performances du dispositif marqué CE équivalent ne sont pas suffisantes pour le groupe de patients cible
- Les établissements de santé/laboratoires doivent respecter l'article 5, paragraphe 5, et les LDT doivent répondre aux exigences générales de sécurité et de performance (annexe I) de l'IVDR
- Les États membres de l'UE superviseront les dispositifs LDT fabriqués et utilisés dans les établissements de soins de santé sur leur territoire
Règles pour les établissements de santé
Quelles sont les principales exigences pour les institutions scientifiques souhaitant fabriquer et utiliser des LDT ?

Fabrication et utilisation des LDT - Exigences clés pour les établissements de santé
Les LDT doivent être fabriqués et utilisés dans le cadre d'un système de gestion de la qualité
L'établissement de santé doit être accrédité conformément à la norme EN ISO 15189 (ou à d'autres dispositions nationales)
Les LDT doivent être conformes aux exigences générales de sécurité et de performance (GSPR - Annexe I de l'IVDR)
L'établissement de santé examinera l'expérience acquise lors de l'utilisation clinique et prendra les mesures correctives nécessaires, le cas échéant
L'établissement de santé compilera la documentation sur la fabrication, la conception et les performances des dispositifs
L'établissement de santé rendra public la déclaration de conformité

Exigences en matière de documentation conformément à l'article 5, paragraphe 5
Les établissements de santé doivent préparer une documentation qui décrit :
- La finalité du dispositif
- L'installation de fabrication
- Le processus de fabrication
- Les données relatives à la conception et aux performances de l'appareil au regard de sa finalité
La documentation doit être suffisamment détaillée pour démontrer que tous les GSPR applicables, tels que décrits à l'annexe 1, sont respectés
L'IVDR prescrit l'exigence ci-dessus pour les dispositifs de la classe D. Toutefois, le règlement permet aux États membres d'appliquer cette disposition également aux dispositifs de classe A, B ou C.
VOTRE LABORATOIRE EST-IL PRÊT POUR L'IVDR ?
PRENEZ CONTACT AVEC NOUS DÈS AUJOURD'HUI !
Les établissements de santé et la préparation à l'IVDR
La préparation est la clé !
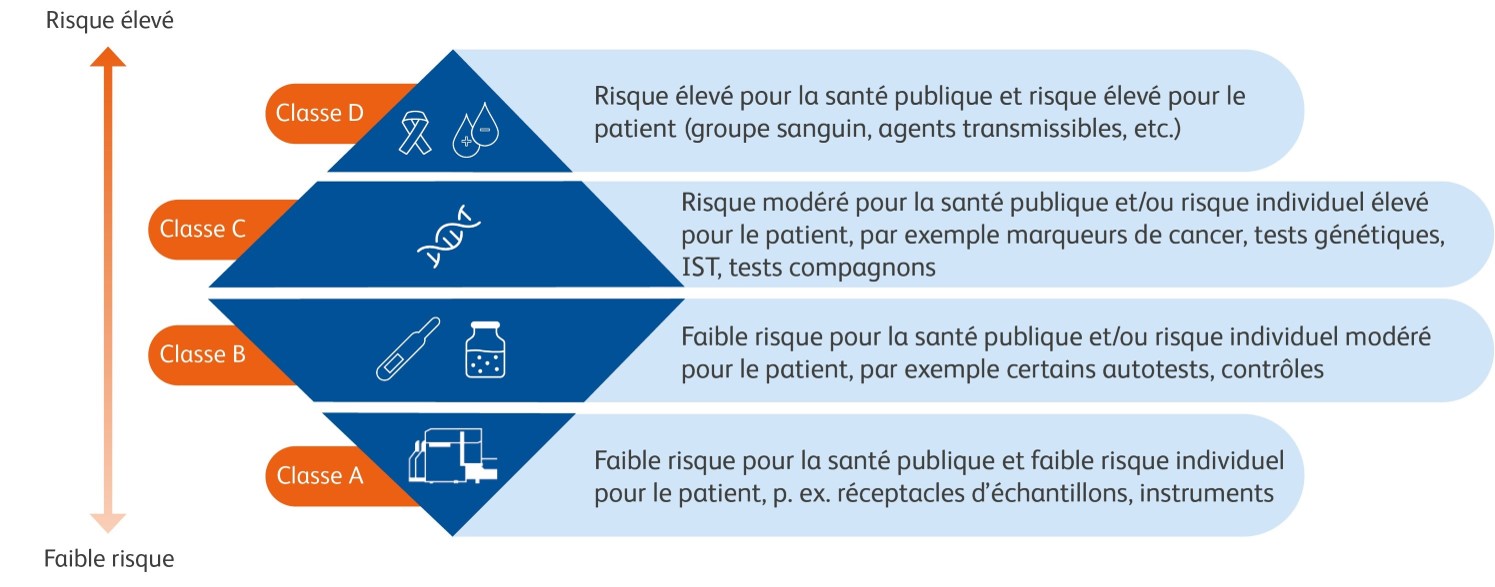
Nouvelles classifications et exigences dans le cadre de l'IVDR
Nouveau système de classification basé sur le risque
Nouvelle évaluation de la conformité
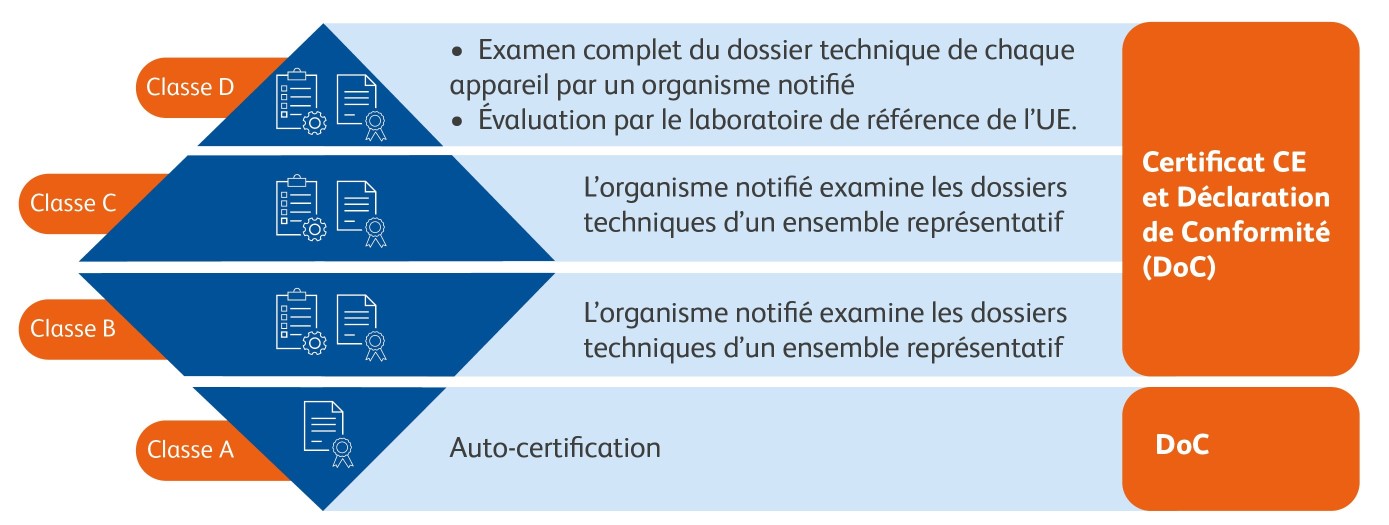
Pour les fabricants, l'IVDR prévoit une procédure d'évaluation de la conformité pour chaque classe de risque comme condition préalable à la déclaration de conformité, le cas échéant.
Il existe des exigences supplémentaires pour les classes C et D
Rapports pour chaque classe d’appareil
- Résumé de la sécurité et des performances (SSP)
- Rapport périodique de mise à jour de la synthèse
Evaluation de la surveillance
- Annuelle, par l'organisme notifié pour garantir que les fabricants appliquent le système de gestion de la qualité (QMS) et le plan de surveillance post-commercialisation (PMS) approuvés
- L'organisme notifié peut rendre visite aux fournisseurs ou sous-traitants essentiels
Audits non annoncés par un organisme notifié
- Au moins tous les 5 ans
- L'organisme notifié peut tester les dispositifs sur place et/ou sur le marché
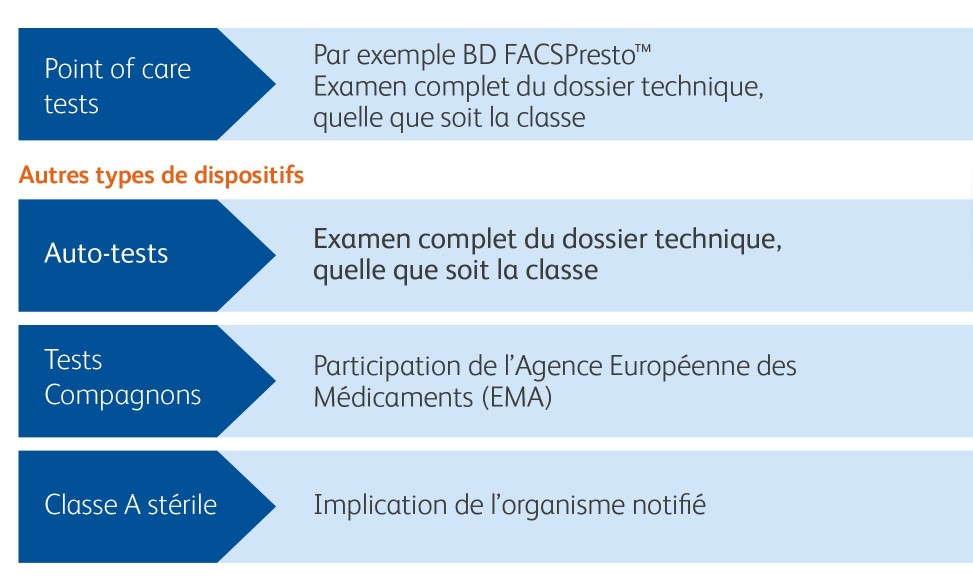
Considérations supplémentaires pour des types de dispositifs spécifiques
Nouvelle évaluation de la conformité pour des types de dispositifs spécifiques

Traçabilité et Transparence
Comment l’IVDR vise à atteindre ces objectifs
Traçabilité et transparence
Amélioration des niveaux de transparence et de traçabilité

Le marquage CE

Déclaration de conformité
- Est la base du marquage CE
- Il s'agit du document dans lequel le fabricant déclare la conformité des dispositifs à toute la législation applicable en matière de marquage CE.
Nouvelles exigences pour le document DoC par rapport à l'IVDD :
- Comprend le SRN - Numéro d'enregistrement unique*.
- Comprend le BUDI-DI
- Traduction dans une langue officielle de l'UE requise par les États membres
Traduction dans une langue officielle de l'UE requise par les États membres
Vous souhaitez échanger avec BD autour de l’IVDR ? Contactez-nous !


