
EU IVDR
NÄCHSTE EPISODE
Hier finden Sie sämtliche vorhergehenden Folgen, sollten Sie welche verpasst haben!
Folge 2
Folge 1
Trailer
Sie möchten nichts versäumen?
Füllen Sie einfach dieses Formular aus
Meistern Sie die Herausforderung durch die schwierigen Gewässer der Europäischen Verordnung zu In-vitro-Diagnostika (2017/746) zu navigieren.
Die Europäische Verordnung 2017/746 zu In-vitro-Diagnostika (EU-IVDR) stellt für uns alle eine Herausforderung dar. Doch gibt es immer einen Weg, eine solche zu bewältigen!
Wir haben die Chance, uns dieser großen Herausforderung gemeinsam zu stellen und partnerschaftlich zusammenzuarbeiten, damit wir den Wechsel zur IVDR meistern und ihre Anforderungen erfüllen.
Die positiven Veränderungen, die die Europäische Verordnung für In-vitro-Diagnostika mit sich bringt, versetzt uns weiterhin in die erfreuliche Lage, Ihnen und Ihren Patienten unsere spezifischen Produkte und Technologien zur Verfügung zu stellen.
Was bedeutet die EU-IVDR für medizinische Einrichtungen?
Die EU-IVDR betrifft alle Medizinprodukte zu In-vitro-Diagnostika und bringt verschiedene Veränderungen mit sich. Hersteller müssen ihr Produktportfolio überprüfen und eventuell bestimmte Produkte aus dem Verkehr ziehen.
Medizinische Einrichtungen sollten wissen, welche ihrer Produkte betroffen sind und entsprechende Vorbereitungen treffen, um ab dem Geltungsdatum der IVDR Kontinuität gewährleisten zu können.
Auch vom Labor entwickelte Tests (Lab Developed Tests, LDT) fallen unter die IVDR. Das bedeutet, dass ein LDT bestimmte Anforderungen erfüllen muss und die medizinische Einrichtung den LDT nur dann anwenden darf, wenn diese erfüllt sind.
Planung und Vorbereitung sind entscheidend!
Was beinhaltet die IVDR?

Einführung
Einführung Die Europäische Verordnung zu In-vitro-Diagnostika 2017/746 (IVDR) trat am 26. Mai 2017 in Kraft – zunächst mit einer Übergangsphase von 5 Jahren, die bis zum 26. Mai 2022 gilt.
Die IVDR ersetzt die aktuelle EU-Richtlinie 98/79/EG zu In-vitro-Diagnostika (IVDD), mit dem Ziel die Gesundheit zu verbessern, mehr Sicherheit für die Bereitstellung und Verwendung von Produkten auf dem EU-Markt zu gewährleisten und neue Regeln für die CE-Zertifizierung von In-vitro-Diagnostika zu etablieren. Bei der IVDR handelt es sich um eine Verordnung, die nicht in der nationalen Gesetzgebung verankert werden muss, weshalb sie eine einheitliche Interpretation in der gesamten EU sicherstellt.
Ziele der IVDR
Patientensicherheit
um ein hohes Maß an Gesundheit für die Menschen und mehr Schutz für die Patienten gewährleisten.
Freier & fairer Handel
um eine problemlose Funktion des Binnenmarktes sicherzustellen
Innovation
um den rechtlichen Rahmen bereitzustellen, der Innovation und Wettbewerbsfähigkeit des europäischen Medizinproduktemarktes unterstützt
Harmonisierung
Die Verordnung muss von allen Mitgliedsstaaten vollständig umgesetzt werden.
Vom Labor entwickelte Tests
Welche Auswirkungen hat die IVDR auf Tests, die vom Labor entwickelt werden (Lab Developed Tests, LDT)?

Warum gehören LDT zum Anwendungsbereich der neuen IVDR?
LDT unterliegen von nun an der IVDR. Die Regeln für die Anwendung und Herstellung von LDT sind jetzt durch die IVDR festgelegt, um ein Höchstmaß an Schutz sicherzustellen – einem der Hauptziele der IVDR.
In der jetzigen EU-Richtlinie zu In-vitro-Diagnostika (IVDD 98/79/EG) werden keine Anforderungen an LDT beschrieben.

Medizinische Einrichtungen (ME), die ihre LDT verwenden möchten, müssen diese Regeln berücksichtigen
- Die Entwicklung und Anwendung eines LDT ist nur dann möglich, wenn kein CE-zertifiziertes Alternativprodukt vorliegt oder die Leistung des entsprechenden CE-zertifizierten Produkts für die spezifische Patientengruppe unzureichend ist.
- ME/Labore müssen sich gemäß Artikel 5(5) verhalten, und der LDT muss die allgemeinen Sicherheits- und Leistungsanforderungen (Anhang I) der IVDR erfüllen
- Die EU-Mitgliedstaaten werden die LDT-Produkte, die in ihren medizinischen Einrichtungen hergestellt und verwendet werden, überwachen
Regeln für medizinische Einrichtungen
Zentrale Anforderungen für medizinische Einrichtungen

Zentrale Anforderungen für medizinische Einrichtungen bezüglich der Herstellung und Anwendung von LDT
Der LDT muss im Rahmen eines Qualitätsmanagementsystems hergestellt und angewendet werden
Die medizinische Einrichtung muss entsprechend EN ISO 15189 (oder alternativen nationalen Bestimmungen) akkreditiert sein
Der LDT muss den allgemeinen Sicherheits- und Leistungsanforderungen (General Safety and Performance Requirements, GSPR) im Anhang I der IVDR entsprechen
Die medizinische Einrichtung überprüft ihre Erfahrungen mit dem LDT, die sie durch die klinische Anwendung gewonnen hat, und ergreift, falls erforderlich, korrektive Maßnahmen
Die medizinische Einrichtung erstellt die Dokumentation zu Herstellung, Design und Leistungsfähigkeit des Produkts
Die medizinische Einrichtung macht der Öffentlichkeit eine Konformitätserklärung zugänglich

Dokumentationsanforderungen gemäß Artikel 5(5)
Die Dokumentation medizinischer Einrichtungen sollte folgende Punkte beinhalten:
- Den Verwendungszweck des Produkts
- Die Herstellungsstätte
- Den Herstellungsprozess
- Design- und Leistungsdaten des Produkts in Hinblick auf seinen Verwendungszweck
Die Dokumentation sollte ausreichend detailliert sein und verdeutlichen, dass alle anwendbaren GSPR gemäß Anhang I der IVDR erfüllt werden.
Gemäß IVDR ist die oben genannte Anforderung für Klasse-D-Produkte vorgeschrieben. Jedoch gestattet die Verordnung den Mitgliedsstaaten auch, sie für Klasse-A-, Klasse-B- oder Klasse-C-Produkte anzuwenden.
Gesundheitsinstitutionen und Bereitschaft für die IVDR
Vorbereitung ist der Schlüssel zum Erfolg
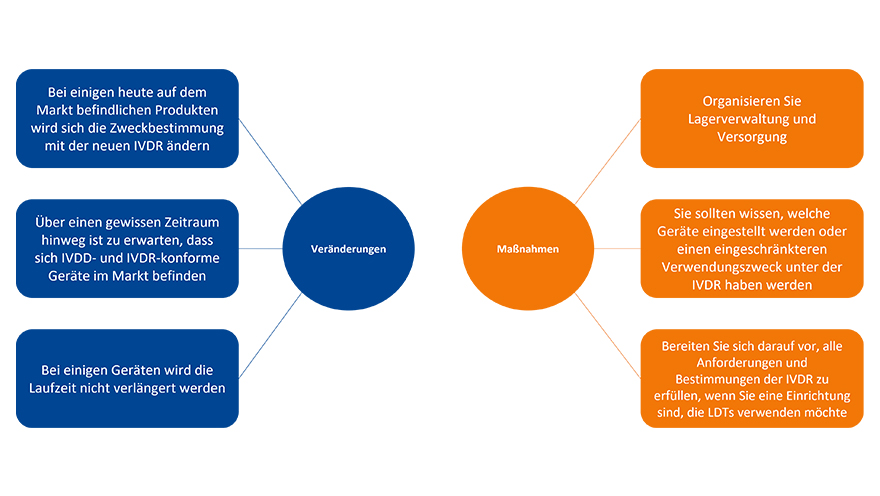
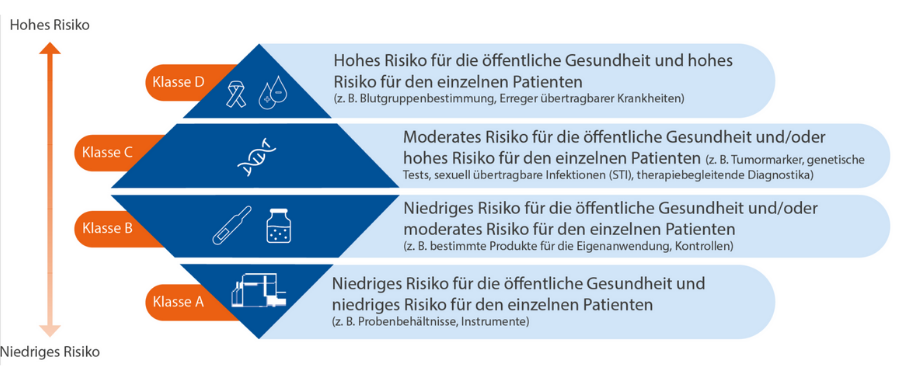
Neue Klassifizierung und Anforderungen der IVDR
Neues risikobasiertes Klassifikationssystem
Neue Konformitätsbewertung
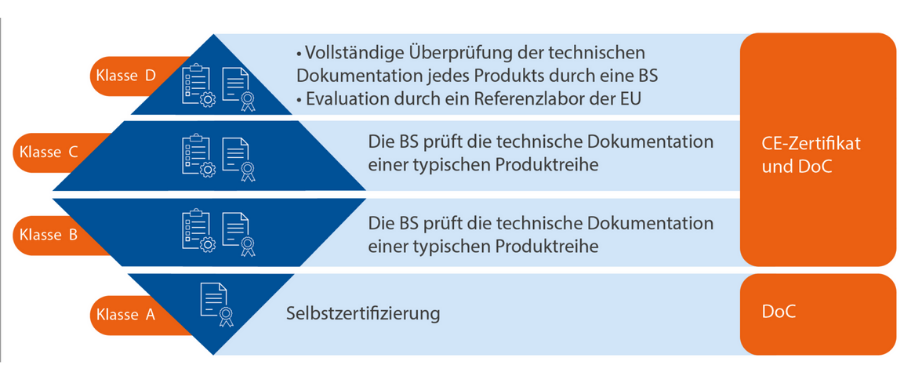
BS (Benannte Stelle) = Eine Benannte Stelle ist ein unabhängiges Zertifizierungsorgan, das Konformitätsbewertungen für die Produkte durchführt, die nicht selbst zertifiziert werden. Nach Bestätigung der Regelkonformität wird das CE-Zertifikat von der Benannten Stelle ausgestellt. Anschließend kann der Hersteller die Konformitätserklärung (Declaration of Conformity, DoC) ausfertigen. Die DoC bestätigt, dass alle maßgeblichen gesetzlichen Anforderungen zur CE-Zertifizierung der aufgelisteten Produkte erfüllt werden.
Für Hersteller sieht die IVDR ein Konformitätsbewertungsverfahren für alle Risikoklassen vor. Dieses ist Voraussetzung für die Konformitätserklärung (DoC) und, wo anwendbar, für das CE-Zertifikat.
Weitere Anforderungen für die Produktklassen C und D
Für die Produktklassen C und D bestehen weitere Anforderungen
Berichte für jede Produktklasse
- Zusammenfassung der Sicherheit und Leistungsfähigkeit (Summary of Safety and Performance, SSP)
- Regelmäßiger zusammenfassender Aktualisierungsbericht (Periodic Summary Update Report, PSUR)
Überwachungsbewertung
- Jährlich durch eine Benannte Stelle, um sicherzustellen, dass Hersteller das zugelassene Qualitätsmanagementsystem (QMS) und den Plan zur Marktbeobachtung (Post-Market-Surveillance, PMS) berücksichtigen
- Die Benannte Stelle kann kritische Lieferanten oder Unterauftragnehmer besuchen
Unangekündigte Prüfungen durch eine Benannte Stelle
- Mindestens alle 5 Jahre
- Die Benannte Stelle kann Produkte vor Ort und/oder vom Markt prüfen
Weitere Prüfungen für spezifische Produktarten
Neue Konformitätsbewertung für spezifische Produktarten


Nachverfolgbarkeit und Transparenz
Gehören zu den zentralen Aspekten, um die IVDR-Ziele zu erreichen
Verbesserte Transparenz und Nachverfolgbarkeit mit der IVDR

Die CE-Zertifizierung

Konformitätserklärung (DoC)
- Ist die Basis für eine CE-Zertifizierung
- Ist das Dokument, in dem der Hersteller die Konformität der Geräte mit allen geltenden Rechtsvorschriften für die CE-Kennzeichnung erklärt
Neue Anforderungen für das DoC-Dokument im Vergleich zur IVDD:
- beinhaltet die Single Registration Number* (SRN)
- beinhaltet die Basic UDI-DI (BUDI-DI)
- wird gemäß Forderung der Mitgliedsstaaten in eine offizielle EU-Sprache übersetzt
* Die SRN ist eine einmalige Registrierungsnummer, die dem Wirtschaftsbeteiligten (Hersteller, bevollmächtigter Vertreter, Importeur) nach der Registrierung in der EUDAMED zugewiesen wird.


